The PRIMEmodel repository provides machine learning-based tools for the genome-wide prediction of regulatory elements using CAGE and other 5’-end sequencing data. The core model, based on LightGBM, was trained on GM12878 whole-cell CAGE and nucCAGE data from the Andersson lab. Available as an R package, PRIMEmodel automates the entire analytical workflow, from processing input bigWig files to the accurate identification of enhancers and promoters. To maximize user flexibility, the tool offers two primary modes of operation:
- R package: Features two main functions that encapsulate the complete pipeline for streamlined, user-friendly execution.
- Bash scripts: Provides pre-configured scripts to execute the pipeline in its entirety or in modular parts. This option is ideal for users who prefer a command-line interface or require automation for large-scale data processing tasks.
Additionally, individual steps can be run independently or seamlessly integrated via a master bash script for selective execution. Although the project was initially developed and optimized for the human reference genome (hg38), the pipeline can be readily adapted for use with other species.
Main Concept of PRIMEmodel for Genomewide Prediction
- The main PRIMEmodel workflow [running with ./PRIMEmodel.sh –predict]
The workflow focuses on the prediction of regulatory elements using machine learning, encompassing six key steps: 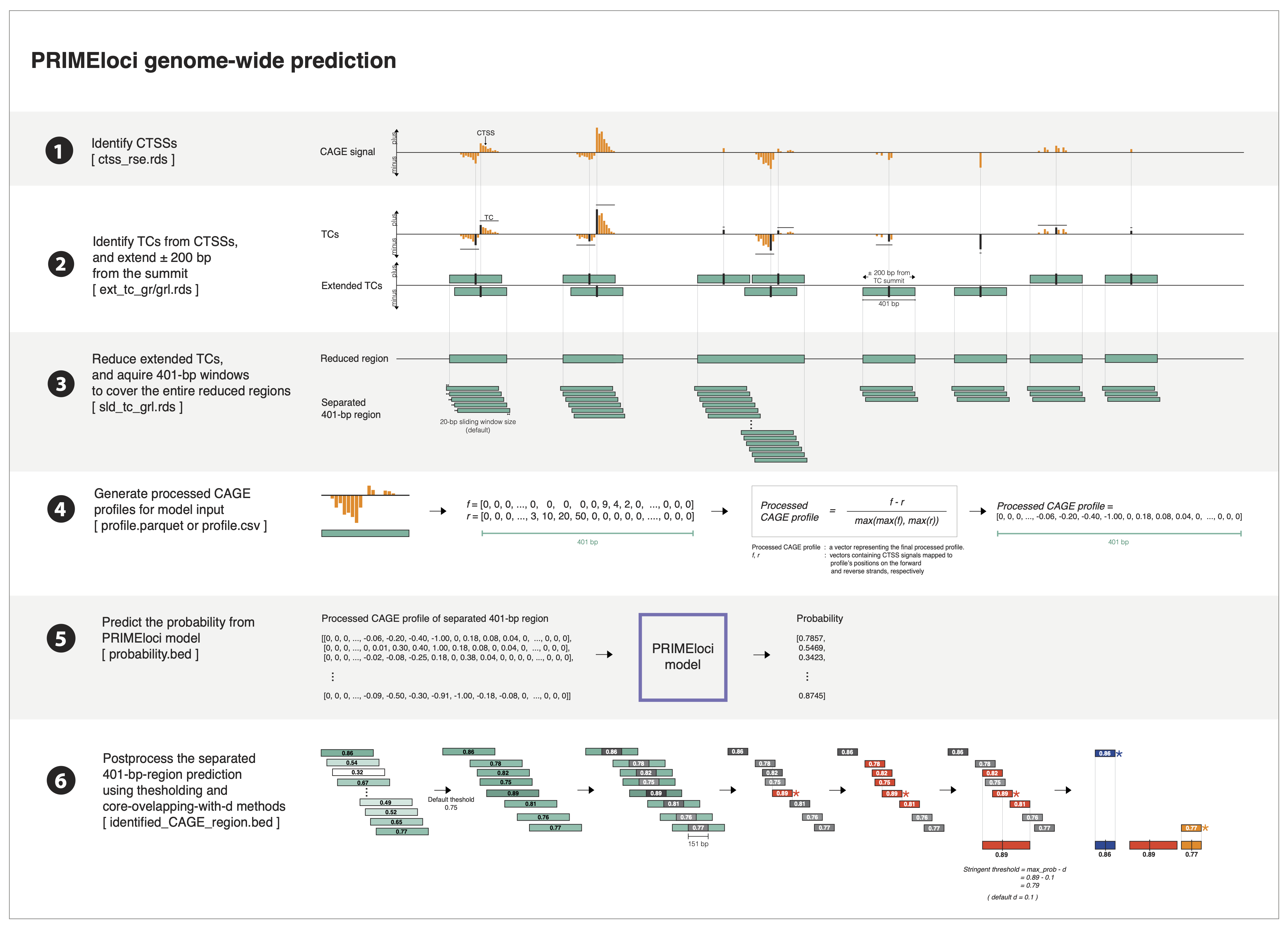
1) Extracting CTSS Data : Extract CAGE transcriptional start site (CTSS) objects from bigWig files using the CAGEfightR package.
2) Identifying Tag Clusters (TCs) : Identify tag clusters (TCs) from the extracted CTSS data using the CAGEfightR package.
3) Sliding Through TCs : Slide through the identified TCs, default setting the window size to 20, to prepare data for downstream analysis.
4) Creating Normalized Profiles : Generate normalized profiles for input into the prediction model.
5) Predicting Profile Probabilities : Use PRIMEmodel models to predict the probabilities of regulatory elements.
6) Post-Processing : Refine and filter predictions using additional criteria for improved accuracy, outputting non-overlapping loci in .bed format for further analysis in R.
Each of these steps can be run separately, and recommended running it separately for the larger dataset (either large number of libraries or very high sequencing depth). All intermediate files here will be kept as it may be used later on for the downstream analysis.
-
PRIMEmodel focal workflow [running with ./PRIMEmodel.sh –predictFocal]
This workflow is designed to use prediction results generated by PRIMEmodel or from fixed genomic regions. The input consists of:
CTSS data stored in a
RandomizedSumExperimentobject (ctss_rse.rds)-
Genomic regions defined as a
GRangesobject (region.rds)These CTSS signals are mapped to the specified regions, and normalized profiles (step -4) is generated. Predictions (step -5) are then made for each region across all libraries.
This workflow was developed as part of running PRIMEmodel across FANTOM5 project CAGE data and is applicable to a variety of use cases. Briefly, the project began by calling CTSS using CAGEfightR and pooling all libraries from the FANTOM5 database. After applying light filtering, the data were saved as an
.rdsfile. PRIMEmodel steps 2–6 were then executed on the pooled data to generate prediction outputs. Subsequently, step -7 was used to convert the pooled predictions into aGRanges.rdsfile representing the predicted genomic regions. These pooled predicted regions were then used as input for the focal PRIMEmodel analysis. In this step, CTSS data from each individual FANTOM5 library (not the pooled one) and the pooled predicted regions were used to run focal PRIMEmodel, producing cell-facet-level data for further analysis.
The resulting FANTOM5 PRIME atlas predictions are available on Zenodo.
Installation
To install the PRIMEmodel R package: - R ≥ 4.4 is recommended (PRIMEmodel was developed and tested under R 4.4). - R ≥ 4.2 is also possible, but may require additional installation steps. - Python ≥ 3.9 is required.
PRIMEmodel is implemented based on two other R packages: - CAGEfightR (https://bioconductor.org/packages/devel/bioc/vignettes/CAGEfightR/inst/doc/Introduction_to_CAGEfightR.html) - PRIME (https://github.com/anderssonlab/PRIME)
Please make sure both are installed before installing PRIMEmodel.
The complete installation procedure can be found here: https://github.com/anderssonlab/PRIMEmodel/blob/main/install_PRIMEmodel.md
Usage of Bash Script
To use PRIMEmodel, follow these steps:
-
Navigate to the Genome-wide Prediction Directory (or else run the full path to the PRIMEloc.sh script)
-
Configure Parameters
This workflow is designed to use a configuration file to ensure that all parameters are explicitly defined, helping to prevent confusion around file paths and settings across different steps. Example configuration files are available at:
path/to/PRIMEmodel/genomewide_prediction/bash_config_PRIMEmodel_predict.shpath/to/PRIMEmodel/genomewide_prediction/bash_config_PRIMEmodel_predictFocal.sh
To run the workflow, provide a valid configuration file using the
--configoption. This file must specify all required parameters for the script. The included example settings are compatible with CAGE files located inPRIMEmodel/example/resources. -
Run the Scripts
Execute the desired steps with the following options:
-
Run PRIMEmodel and PRIMEmodel_focal:
./PRIMEmodel.sh --config <config_file> --predict ./PRIMEmodel.sh --config <config_file> --predictFocal # example <config_file> ./PRIMEmodel.sh --config bash_config_PRIMEmodel_predict.sh --predict ./PRIMEmodel.sh --config bash_config_PRIMEmodel_predictFocal.sh --predictFocalNote that if you prefer to run the full workflow in small samples or if server storage is limited, it is recommended to execute the workflow directly within R. This approach runs all steps sequentially and automatically cleans up intermediate files to reduce storage usage.
-
Run individual steps
Each step corresponds to a specific script or function:
-0: Validation CTSS data and given regions-1: Extract CTSS data-2: Identify tag clusters-3: Slide through TCs with a specific window size-4: Create normalized profiles-5: Predict regulatory element probabilities-
-6: Apply post-processing to refine predictions This step structure can also be used flexibly as needed. For example, if PRIMEmodel step -1 (CTSS calling) has already been completed externally and CTSS data are available, you can start directly from step -2 and continue with steps -3, -4, -5, and -6.
Each step is designed to run independently, allowing users to execute one step at a time and stack the outputs sequentially. This modular design is useful for customizing the workflow, reusing intermediate results, or managing large datasets step by step.
Note that The PRIMEmodel workflow consists of steps -1 to -6, each corresponding to an individual script. The PRIMEmodel_focal workflow includes steps -0, -4, and -5, which are designed to be run on individual libraries using the predicted regions.
-
Run extra-step preparation for PRIMEmodel focal We prepare the automatic script for creating the RDS file for the PRIMEmodel focal input:
bash Rscript _7_prepare_postprocessed_bed_for_focal_input.r --bed_path /path/to/bed/file.bed